
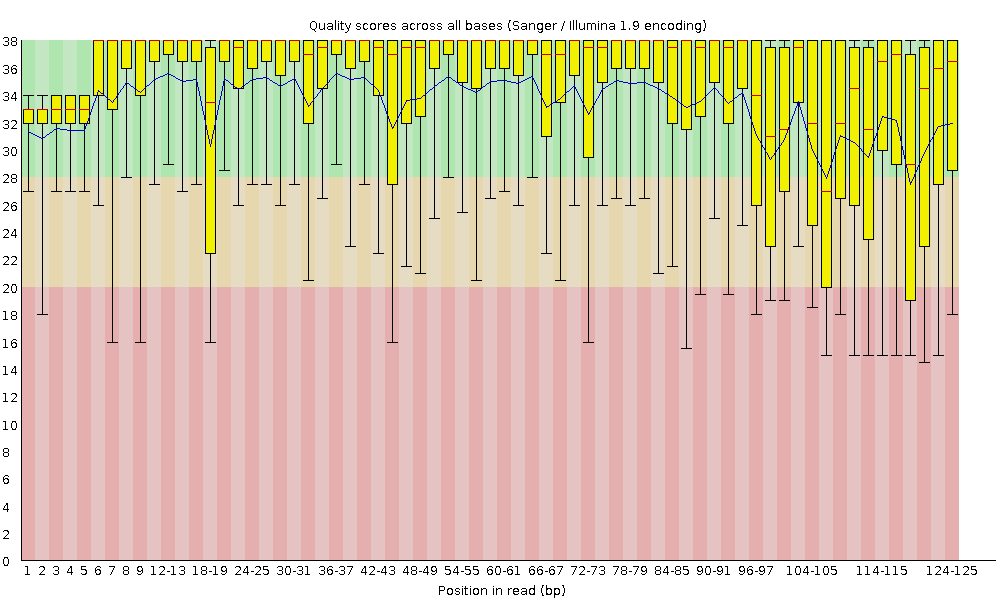
High-throughput sequencing technologies have transformed genomics by enabling massive data generation in record time. But with large volumes of data comes the responsibility of ensuring its quality. This is where FastQC steps in—offering a robust quality control solution tailored for sequencing data.
🔍 What is FastQC?
FastQC is a Java-based application designed to analyze sequencing files (FastQ, BAM, or SAM) and generate comprehensive quality reports. It can operate in:
- Interactive mode, ideal for manual inspection.
- Non-interactive mode, perfect for automated pipelines.
✅ Key Features
- Supports FastQ, BAM, and SAM file inputs.
- Generates HTML reports and summary statistics.
- Works offline and can be integrated into pipelines.
- Built with Picard BAM/SAM Libraries (bundled).
- Open-source under GPL v3 or later.
⚙️ Installation
🔧 Step 1: Install Java Runtime Environment (JRE)
FastQC requires a 64-bit JRE. Install it using your system’s package manager:
Ubuntu / Mint:
sudo apt install default-jre
CentOS / Redhat:
sudo yum install java-1.8.0-openjdk
Check Java installation:
java -version
Expected output:
openjdk version "11.0.2" 2019-01-15
OpenJDK Runtime Environment (build 11.0.2+9)
📦 Step 2: Download and Install FastQC
- Visit the official Babraham Bioinformatics FastQC page.
- Download the version for your operating system.
- Extract the package to your desired directory.
- Make the binary executable (Linux only):
chmod +x fastqc
- Launch with:
./fastqc
🚀 Quick Start Guide
Running a basic analysis with FastQC is simple:
fastqc data.fastq
It generates:
- An HTML report viewable in any browser.
- A .zip archive with the raw output.
🔧 Common FastQC Commands
🔄 Analyze Multiple Files
fastqc file1.fastq file2.fastq file3.fastq
📁 Set Output Directory
fastqc -O /path/to/output/ data.fastq
📦 Skip ZIP Creation
fastqc --noextract data.fastq
🔄 Use Multiple Threads
fastqc -t 4 file1.fastq file2.fastq
⚙️ Disable Interactive Grouping
fastqc --nogroup data.fastq
For more commands, run:
fastqc -h
🧪 Why Use FastQC?
FastQC ensures sequencing data quality before downstream analysis, preventing errors and saving time. Whether you’re:
- Preprocessing RNA-Seq data,
- Running whole-genome sequencing,
- Building automated pipelines,
FastQC is indispensable for validating read quality and spotting anomalies early.
📚 Reference
Andrews, S. (Year). FastQC: A Quality Control Tool for High Throughput Sequence Data [Software]. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
💬 Final Thoughts
Thank you for reading! I hope this guide helped you understand how to leverage FastQC for your NGS projects.
Have questions or suggestions? Drop a comment below or connect with me on social media.

Leave a comment