

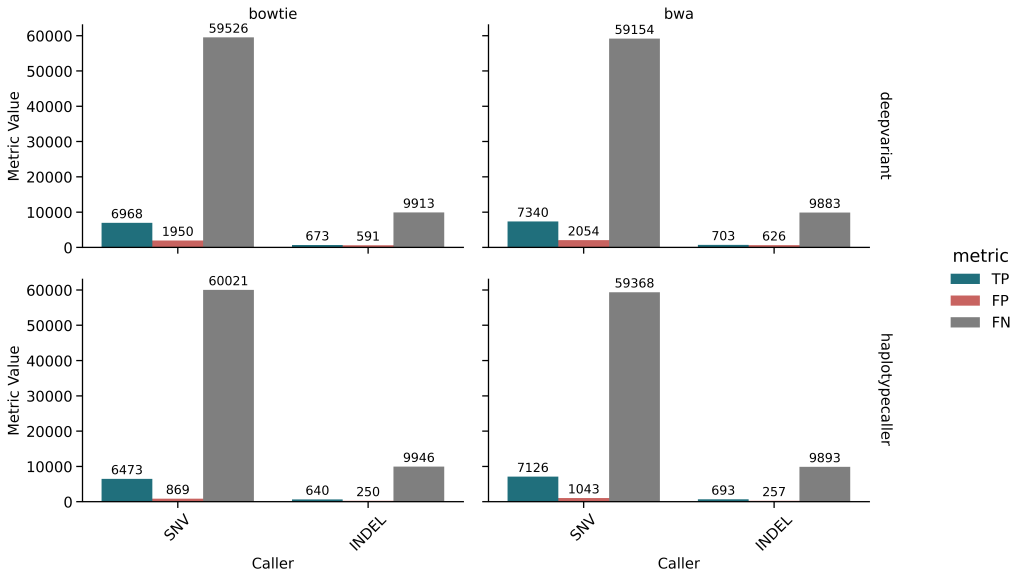
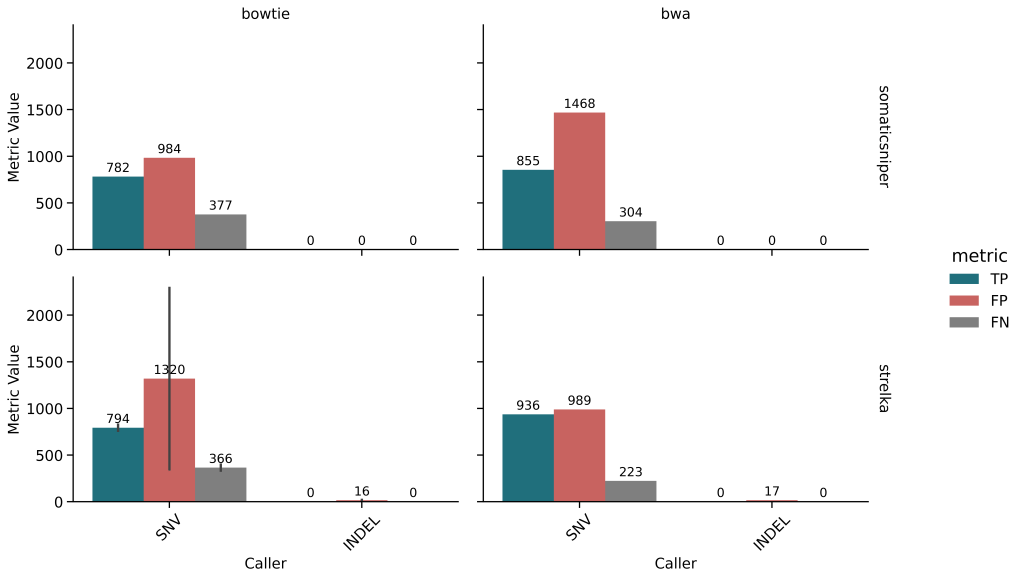
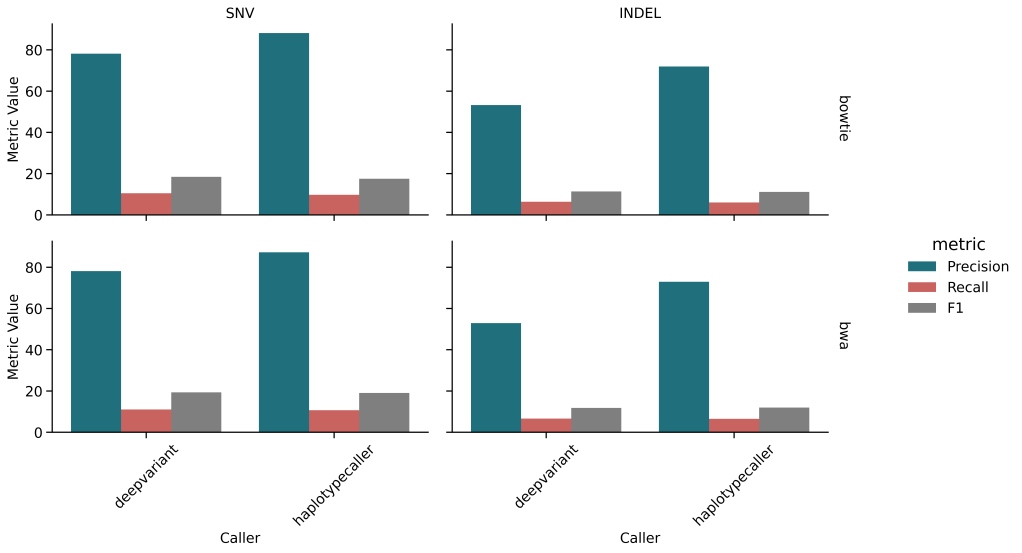
Benchmarking Germline and Somatic Variant Calling Pipelines Using CoSAP and Galaxy
- Course: BLG 555E – Project
- Project Goal: Benchmark the accuracy, reproducibility, and sensitivity of multiple somatic and germline variant calling pipelines across two NGS platforms—CoSAP and Galaxy
- Tools Used: BWA, Bowtie2, Strelka, DeepVariant, HaplotypeCaller, SomaticSniper, GATK, bcftools, snpfilter.pl
- Duration: 6 weeks
- Outcome: Delivered a comprehensive comparison of 9 variant calling pipelines, identified best practices in somatic/germline workflows, and highlighted the critical impact of preprocessing steps on accuracy
A Comparative Study with 9 Pipelines on Real Exome Data
Abstract
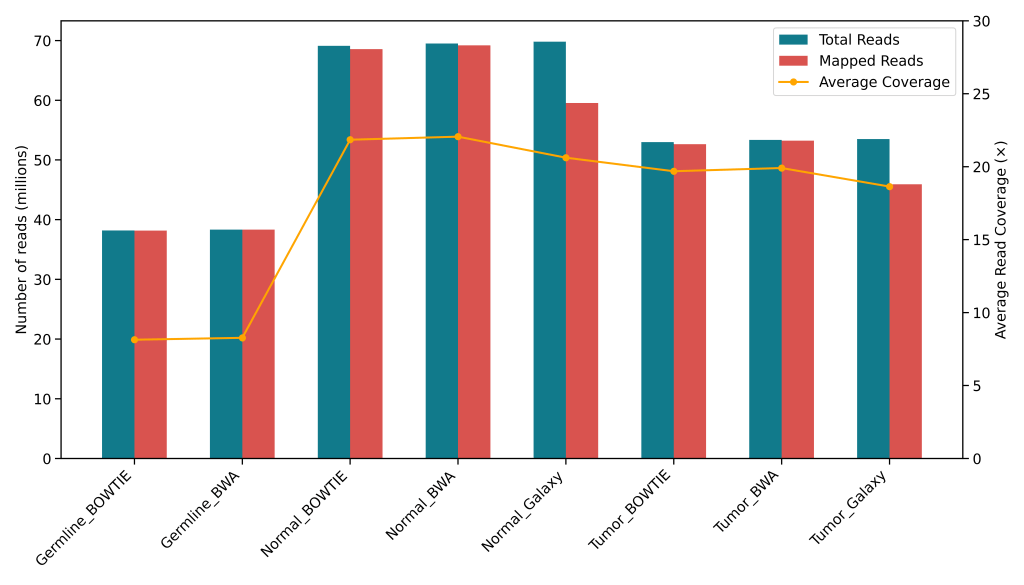
This project evaluated nine variant calling pipelines using whole exome sequencing (WES) data. It involved combinations of two mappers (BWA, Bowtie2) and four variant callers (DeepVariant, HaplotypeCaller, Strelka, SomaticSniper) across both germline and somatic contexts, implemented using CoSAP and Galaxy. Each pipeline was assessed across three quality control layers—FASTQ, BAM, and VCF—using real tumor-normal pairs and a high-confidence germline truth set.
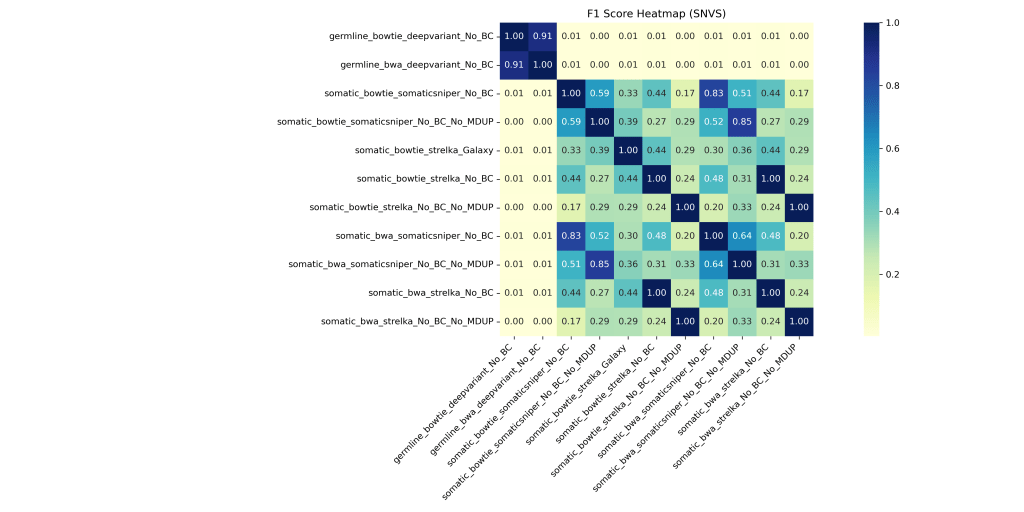
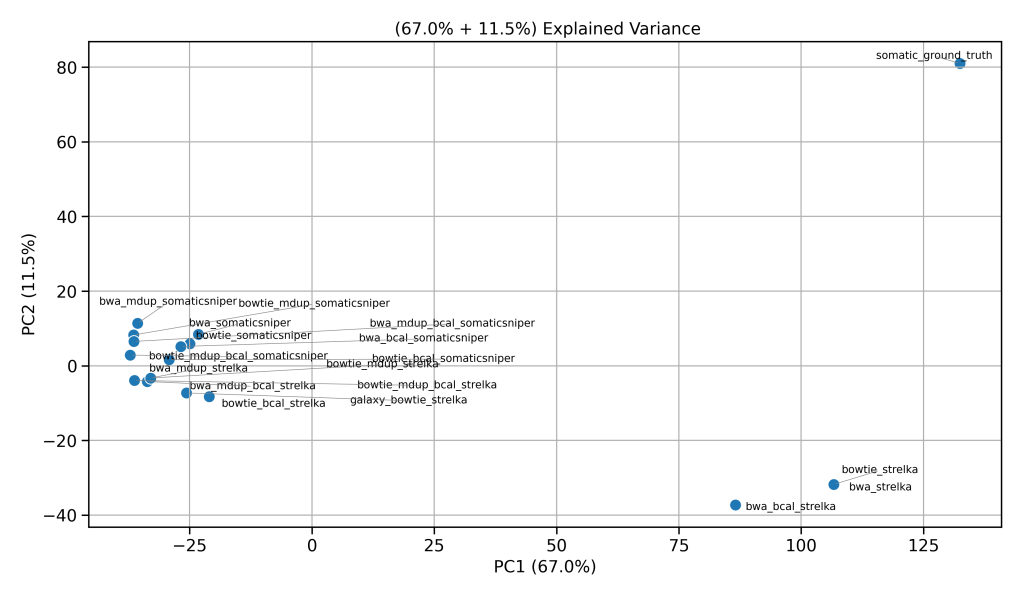
Performance metrics including precision, recall, F1-score, and Jaccard similarity were analyzed, alongside visualizations like UpSet plots, PCA, and heatmaps. The results revealed deep trade-offs in sensitivity vs. specificity and emphasized the importance of trimming, duplicate marking, and quality score recalibration in pipeline performance.
🧠 Problem Statement
NGS variant calling pipelines are diverse, but little guidance exists on which combinations perform best across clinical and research use cases. This study systematically compares key configurations in:
- Somatic mutation detection
- Germline variant calling
- Platform-dependent effects (e.g., Galaxy vs. CoSAP)
The primary research questions:
- How do mappers (BWA vs. Bowtie2) affect downstream variant detection?
- Which variant callers offer the best precision/recall balance?
- Do preprocessing steps (e.g., duplicate marking) significantly alter accuracy?
🗂 Data & Workflow
📌 Datasets:
- Somatic Data: ENA tumor/normal FASTQ (SRR7890850/51)
- Germline Data: GIAB dataset (NIST7035 R1/R2 paired-end reads)
- Truth Sets: Ground truth VCF + BEDs for confident/exonic regions (hg38)
🛠 Pipeline Steps:
- Trimming → Alignment (BWA/Bowtie2)
- Duplicate marking and/or base recalibration
- Variant calling (DeepVariant, HaplotypeCaller, Strelka, SomaticSniper)
- Filtering using BEDs
- Evaluation with bcftools, custom Python scripts, and visualization tools
💻 Hardware:
- Ubuntu 24.04, 24-core Intel i9, 64GB RAM, 1TB SSD
- Docker, Conda, CoSAP CLI, Galaxy Web UI
📊 Key Results
🔹 Germline Variant Calling
| Pipeline | Precision | Recall | F1-Score |
|---|---|---|---|
| BWA + DeepVariant | 75.01% | 10.43% | 18.32% |
| BWA + HaplotypeCaller | 85.74% | 10.14% | 18.14% |
| Bowtie + DeepVariant | 75.04% | 9.91% | 17.51% |
| Bowtie + HaplotypeCaller | 86.41% | 9.23% | 16.68% |
- Precision is high, but recall is low (<11%)
- HaplotypeCaller slightly outperformed DeepVariant in FP control
- Mappers had small influence on overall germline calling
🔹 Somatic Variant Calling
| Pipeline | Precision | Recall | F1-Score |
|---|---|---|---|
| BWA + Strelka | 48.20% | 80.76% | 60.37% |
| Bowtie + Strelka | 69.65% | 71.27% | 70.45% |
| BWA + SomaticSniper | 36.81% | 73.77% | 49.11% |
| Galaxy + Strelka (Bowtie) | 24.76% | 65.66% | 35.96% |
- BWA + Strelka had best F1, but Bowtie + Strelka had higher precision
- Galaxy pipelines suffered due to lack of trimming, resulting in poor mapping rates
- Preprocessing (trimming, MDUP, BCAL) critical for somatic pipelines
🔍 Visual Insights
- PCA plots revealed that germline pipelines cluster tightly, but somatic predictions diverge from truth sets
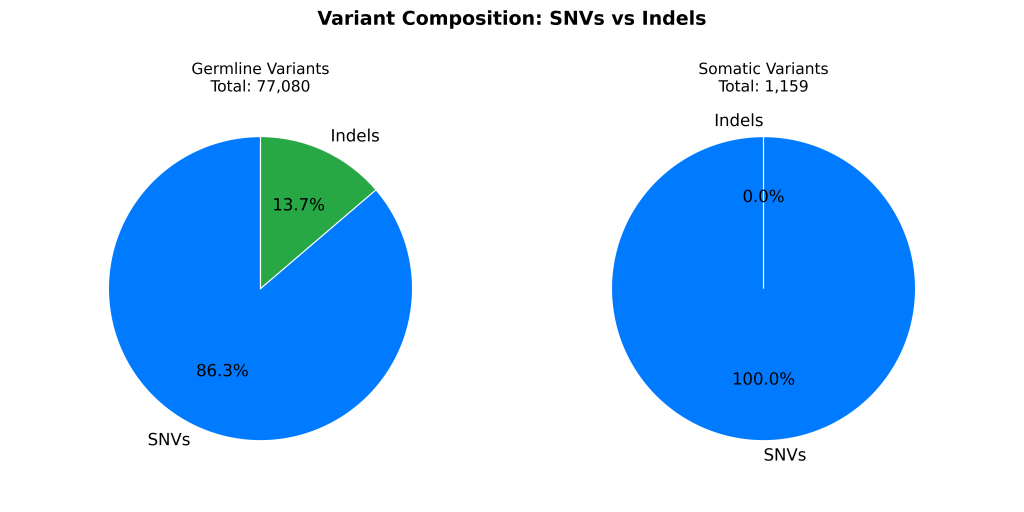
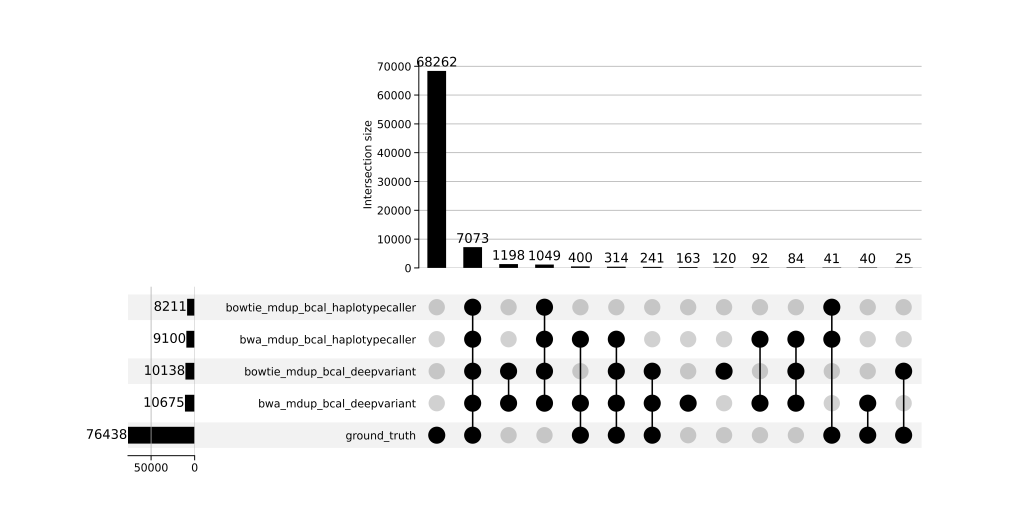
- UpSet plots showed limited overlap between somatic calls and truth (~688 shared variants), while germline pipelines aligned well with >68,000 overlapping variants
- Jaccard similarity: Germline pipelines shared >87% similarity; somatic pipelines ranged 0.28–0.66
- Figure 9 showed that duplicate marking improves F1 scores more than base quality recalibration
💬 Discussion
This benchmarking revealed that:
- Somatic variant calling is more variable and tool-sensitive than germline
- Strelka (especially with BWA) offers the best recall for somatic SNVs
- Germline callers are very precise, but struggle with sensitivity
- Preprocessing steps (especially trimming and duplicate marking) are essential for reliable results
- Galaxy pipelines underperformed due to lack of tuning and preprocessing options
📣 Call to Action
Working on your own NGS data and unsure which pipeline is best?
💬 I can help benchmark, configure, and deploy optimized pipelines for somatic or germline variant calling.
📧 Start a conversation and let’s make your data analysis reproducible and reliable.



Leave a comment